Ознаки та пороки, які визначаються рецесивними генами. Окремим геном і генотипом
рецесивні гени
Рецесивний ген (тобто ознака, їм визначається) може не проявлятися У ОДНОГО АБО БАГАТЬОХ ПОКОЛІНЬ поки не зустрінуться два ідентичних рецесивні гени від кожного з батьків (раптовий прояв такої ознаки у нащадків не слід плутати з мутацією).
Собаки, які мають лише один рецесивний ген - визначник якої-небудь ознаки, які не проявлять це ознака, так як дія рецесивного гена буде замасковано проявом впливу парного йому домінантний ГЕНА. Такі собаки (носії рецесивного гена) можуть бути небезпечні для породи, якщо цей ген визначає появу небажаного ознаки, тому що буде ПЕРЕДАВАТИ ЙОГО СВОЇМ НАЩАДКАМ, а ті далі і він таким чином збережеться в породі. Якщо випадково або необдумано звести в пару ДВОХ НОСІЇВ ТАКОГО ГЕНА вони дадуть частину потомства з небажаними ознаками.
Генетична таблиця, що показує просте взаємодія домінантних і рецесивних характеристик за Менделем
позначення:
А - - домінантний ген від одного з батьків
А -? - рецесивний ген від одного з батьків
АА - - пара домінантних генів, по одному від кожного з батьків
аа -? ? - пара рецесивних генів, по одному від батька
Аа -? - домінантний ген від одного батька і рецесивний - від іншого, складові домінантно - рецесивну пару генів.
Пояснення варіантів:
1. Обоє батьків мають по два домінантних гена, тому всі їх нащадки будуть «чистими» за цією ознакою - все АА.
2. Один батько «чистий» за домінантним фактором (АА), інший тільки виглядає «чистим», але несе рецесивний ген (Аа). Тому всі нащадки будуть виглядати «чистими» від рецесивного ознаки, але насправді половина нащадків буде дійсно «чистої», маючи по два домінантних гена (АА), друга половина буде виглядати «чистої», але кожен з них буде мати рецесивний ген, тобто буде його носієм (Аа). Співвідношення АА: Аа - 1: 1.
3. Один батько зовні проявляє домінантний ознака, але є носієм рецесивного гена (Аа). При спарюванні з партнером, який несе два рецесивні гени і, природно, зовні проявляє ця ознака, половина нащадків буде подібна першому партнеру (Аа), друга - другого. Т.ч., весь послід буде носієм даної ознаки, тільки перші будуть прихованими носіями, а другі - явними. Це особливо важливо розуміти для тих випадків, коли ознака, який визначається рецесивним геном, особливо небажаний для породи.
4. Обоє батьків виявляють домінантний ознака, але кожен несе рецесивний, що не проявляє себе, ген. У посліді буде одна частина нащадків чисто «домінантних», мають два А (АА); дві частини нащадків, зовні виявляючи домінантна ознака і тому, не відрізняючись від перших, приховано нестимуть і рецесивний ознака, маючи Аа; одна частина нащадків проявлятиме рецесивний ознака, маючи два рецесивні гени (аа). Тобто, співвідношення нащадків, які проявляють ознака визначається геном А буде 3: 1, а справжнє розподіл носіїв рецесивного і домінантного генів буде наступним: АА: Аа: аа - 1: 2: 1.
5. У цій парі - один батько «чистий» домінант (АА), інший має два рецесивний гена (аа). Весь послід цієї пари також буде нести цю ознаку, і передавати його нащадкам далі в явній формі до тих пір, поки не будуть спарені з партнером несе домінантний ген. В цьому випадку результат спаровування буде як в парі 3 і 5.
6. Обоє батьків виявляють рецесивний ознака, так як несуть по два рецесивний гена (аа). Весь послід цієї пари також буде нести цю ознаку, і передавати його нащадкам далі в явній формі до тих пір, поки не будуть спарені з партнером несе домінантний ген. В цьому випадку результат спаровування буде як в парі 3 і 5.
Очікуване співвідношення розщеплення нащадків з того чи іншою ознакою приблизно виправдовується при посліді не менше 16 цуценят. Для посліду звичайного розміру - 6-8 щенят - можна говорити лише про більшу чи меншу ймовірність прояву ознаки, що визначається рецесивним геном, для нащадків певної пари виробників з відомим генотипом.
З книги Секрети спадковості людини автора Афонькин Сергій ЮрійовичДомінантні і рецесивні гени Уявіть собі дві гомологічні хромосоми. Одна з них - материнська, інша - батьківська. Копії генів, які працюють на одних і тих же ділянках ДНК таких хромосом називають аллельними або просто алелями (грец. Alios - інший). ці копії
З книги Секрети спадковості людини автора Афонькин Сергій ЮрійовичГени старіння Вік - мерзенна річ, і з кожним роком вона стає все гірше. Діана Купер Люди не хочуть жити вічно. Люди просто не хочуть помирати. Станіслав Лем - Безсмертні клітини існують - Запрограмована загибель - Годинниковий механізм старіння - Хвороби
З книги Еволюція людини. Книга 2. Мавпи, нейрони і душа автораГени і поведінка Факти, здобуті нейробиологами, кажуть про матеріальну, нейрологической природу психіки. Але для того, щоб душа у всіх її проявах могла еволюціонувати, цього, строго кажучи, ще недостатньо. Еволюціонувати можуть не всі ознаки, а тільки
З книги Еволюція людини. Книга 1. Мавпи, кістки і гени автора Марков Олександр ВолодимировичНові гени? Активність генів може змінюватися в ході еволюції не тільки шляхом змін сайтів зв'язування ТФ, роботи самих ТФ або регуляторних РНК, але і в результаті дуплікації генів. За інших рівних два однакових гена зроблять більше продукту (тобто інформаційної
автора Хармар Хіллер З книги і налагодження енергетичного устаткування автора Хармар Хіллер автора Хармар ХіллерРецесивні гени рецесивний ген (т. Е. Ознака, їм визначається) може не проявлятися У ОДНОГО АБО БАГАТЬОХ ПОКОЛІНЬ поки не зустрінуться два ідентичних рецесивні гени від кожного з батьків (раптовий прояв такої ознаки у нащадків не слід плутати з
З книги Собаки і їх розведення [Розведення собак] автора Хармар ХіллерДомінантні гени Присутність домінантного гена завжди явно і зовні проявляється характерною ознакою. Тому домінантні гени, що несуть небажаний ознака, представляють для селекціонера значно меншу небезпеку, ніж рецесивні, так як їх присутність
З книги Собаки і їх розведення [Розведення собак] автора Хармар ХіллерЛетальні гени Це гени, що викликають загибель організму до досягнення нею статевої зрілості. Летальні гени є рецесивними, ось кілька прикладів прояви їх впливу: «заяча губа і вовча паща» - дефект розвитку верхньої щелепи, гемофілія - відсутність у крові
З книги Неандертальці [Історія невдалого людства] автора Вішняцкій Леонід Борисович автора Кандель Ерік Річард З книги В пошуках пам'яті [Виникнення нової науки про людську психіку] автора Кандель Ерік Річард автора Хенгстшлегер МаркусГени спортсмена Жінки люблять його за його зовнішність, а чоловікам подобається його вміння забивати м'ячі. Жінки захоплюються його зачіскою і звабливою посмішкою, чоловіки поважають його за кидки з флангу. Йдеться про двох персон. Після всього, що ми дізналися про генетичне
З книги Влада генів [прекрасна як Монро, розумний як Ейнштейн] автора Хенгстшлегер МаркусГени Моцарта Про дитинство Моцарта не так багато точних відомостей, куди більше - припущень. Як все відбувалося у цього маленького генія? Сестра Наннерль, старша за нього на п'ять років, захотіла дізнатися побільше про свого молодшого брата. Який він був насправді в ранньому дитинстві? В
З книги Влада генів [прекрасна як Монро, розумний як Ейнштейн] автора Хенгстшлегер МаркусГеніальний гени А тепер зовсім інше питання: чому це так цікаво? Що можна такого дізнатися, дослідивши череп Моцарта? Ну, припустимо, його зважили і виміряли. За результатами аналізу кісток можна з'ясувати, що їв власник цього черепа. І це теж вже зробили. цей череп
З книги Внутрішня риба [Історія людського тіла з найдавніших часів до наших днів] автора Шубін НілГени Відкриття Арендта підводить нас до ще одного питання. Одна справа, що у очей різних тварин є спільні частини, але як вийшло, що такі несхожі один на одного очі, як у черв'яків, мух і мишей, близькі одна одній? Щоб відповісти на це питання, звернемося до
спадковість -властивість організмів повторювати в ряді поколінь схожі ознаки. Завдяки спадковості батьки і нащадки мають подібні зовнішність, статура, обмін речовин. Внаслідок цього кожний вид відтворює себе із покоління в покоління.
Наследственностьявляется результатом комбінацій генів.
гени - це біохімічні функціональні елементи хромосом, що визначають потенційний підлогу і інші ознаки у зародка.
хромосоми (Грец. - колір і тіло) - нуклеопротеїдні структури в ядрі еукаріотичної клітини (клітини, що містить ядро), які стають легко помітними в певних фазах клітинного циклу (під час мітозу або мейозу). Хромосоми являють собою високу ступінь конденсації хроматину, постійно присутнього в клітинному ядрі. Початково термін був запропонований для позначення структур, що виявляються в клітині, але в останні десятиліття все частіше говорять про бактеріальних хромосомах. У хромосомах зосереджена велика частина спадкової інформації.
Хромосомипредставлени парами в ядрі кожного сперматозоїда і кожної яйцеклітини. У людини є 23 пари, або 46 хромосом, Причому одну пару називають статевими хромосомами, тому що вони визначають стать майбутньої організму. Клітини жіночого організму несуть дві Х-хромосоми, тоді як клітини чоловічого містять одну Х-і одну Y-хромосому. Y-хромосома дрібніше за розміром і містить менше генів на своїй поверхні, ніж Х-хромосома. В результаті клітинного ділення в яєчниках все яйцеклітини містять одну Х-хромосому, тоді як клітинний розподіл в яєчках призводить до того, що половина сперматозоїдів містить Х-хромосому, а інша їх половина - Y-хромосому. Тобто, в половині випадків людська яйцеклітина запліднюється сперматозоїдами, що несуть Х-хромосому, і ще в половині - сперматозоїдами, що несуть Y-хромосому, так що половина народжуються в популяції дітей повинна б мати чоловічу стать, а половина - жіночий.
домінантний ген(dominant gene) - ген, виражений в фенотипі незалежно від присутності в геномі іншого аллеля цього гена. Присутність якого забезпечує прояв визначається їм ознаки незалежно від того, чи є інший ген тієї ж пари домінантним або рецесивним.
рецесіявний ген(recessive) - генетична інформація, яка може придушуватися впливом домінантного гена. Рецесивний ген здатний забезпечити прояв визначається їм ознаки тільки в тому випадку, якщо знаходиться в парі з відповідним рецесивним геном. Якщо ж він знаходиться в парі з домінантним геном, то він не проявляється, так як домінантний ген придушує його. Властивості, представлені рецесивними генами, виявляються в фенотипі у нащадка лише в тому випадку, якщо в обох батьків є рецесивний ген.
Коли обидва алелі в парі абсолютно однакові (наприклад, ГО, АА), то такий генотип і його володар називаються гомозиготних, а коли ці алелі різні (скажімо, АТ) - гетерозиготних. Відомо, що якщо гомозиготні генотипи ГО і АА зумовлюють першу і другу групи крові відповідно, то у власників гетерозиготного генотипу АТ буде також друга група крові. Це означає, що в такій комбінації проявляється ефект гена А і не проявляється ефект гена О, тобто ген А домінує, А ген Про по відношенню до нього рецессивен (Слово «рецесивний» означає зникаючий). Таким чином, домінантні гени проявляють свою дію як в гомозиготному, так і в гетерозиготному стані, а рецесивні гени можуть проявитися тільки в гомозиготному стані і не дають зовнішніх проявів у гетерозиготних людей.
Сукупність спадкових факторів організму (генів) називається генотипом. Сукупність усіх ознак і властивостей організму, які є результатом взаємодії генотипу і зовнішнього середовища, називаються фенотипом. Тобто фенотип - це відображення генотипу в поєднанні з навколишнім середовищем. Межі в яких навколишнє середовище впливає на прояв генотипу називається нормою реакції.
Джерела інформації:
Загальні дані
Результати взаємодії генів двох батьківських геномів в зиготі і розвиненому з неї багатоклітинних організмі проявляються в контрольованих ними ознаках, які в тій чи іншій мірі передаються з покоління в покоління. Таке спадкування залежить від багатьох причин.
У класичній генетиці тривалий час вважалося, що внесок обох батьків в геном потомства приблизно однаковий як по материнській, так і по батьківській лінії сімейних родоводів. На цій основі сформулювали одне з перших правил спадкування - рівнозначність і взаємопов'язаність функцій двох різних за походженням алелей одних і тих же генів (або еквівалентність реципрокних схрещувань).Виділили два варіанти і ряд типів успадкування з урахуванням кількості генів, їх походження (материнське або батьківське), локалізації в алельних або неалельних локусах аутосом і статевих хромосом, характеру прояву (домінантність або рецесивним), а також особливостей (механізмів) взаємодії між генами.
Проте вже в другій половині XX ст. встановили: у багатьох випадках внесок одного з батьків значно відрізняється від вкладу другого з батьків. Було показано: функції батьківських генів протягом усього онтогенезу можуть змінюватися аж до диференціального відключення материнських або батьківських алелей. В основі цього явища лежить епігеномний процес або маркування локусів хромосом одного з батьків, що приводить до вимикання експресії розташованих в них алелей. Дане явище отримало назву імпринтингу.Термін вперше застосований в другому десятилітті XX в. австрійським зоологом Конрадом Лоренцом. Спостерігаючи за поведінкою каченят, щойно вилупилися з яєчної шкаралупи, він звернув увагу: вони шукають поглядом свою маму-качку. Якщо в поле їх зору потрапляє господар або господиня качки або пробігає повз собака або кішка - за ними, як за своєю матір'ю, каченята йдуть все подальше життя.
Таким чином, мова йде про формування до народження ряду функцій організму, відображених в геномі ( «геномна пам'ять»).
Пізніше термін «імпринтинг» стали застосовувати невропатологи. Импринтингом, вважають вони, обумовлено походження ряду вроджених феноменів у новонароджених немовлят, наприклад таких, як пошук соска материнської грудної залози або базисні функції (ковтання, дихання, жування, кровообіг, травлення і всмоктування), які сформувалися у внутрішньоутробному житті.
Потім цей термін стали використовувати генетики для пояснення незвичайної поведінки статевих хромосом (елімінація хромосом батьківського походження) у комах роду Sciara Coprophila (Кроуз Г., 1960). Було показано: батьківські Х-хромосоми якимось чином маркуються (імпрінтіруются) перед злиттям гамет відповідно до своїх батьківських походженням.
В даний час переконливо доведено общебиологическое значення импринтинга як епігеномний процесу, пов'язаного з регуляторними (функціональними) змінами активності геному і не пов'язаного з його структурними ушкодженнями (див. Розділ 28).
У людини в результаті імпринтингу, наприклад, експресується материнський ген (алель), тоді як батьківський аллель блокується або навпаки. Отже, у індивіда має місце тільки моноаллельная експресія або нееквівалентний внесок в геном одного з його батьків, а значить, в наявності відхилення від менделевских законів успадкування, заснованих на діаллельной моделі (див. Розділ 2).
Перш ніж розглянути такі відхилення, зупинимося на закономірності успадкування генів і ознак, вперше описаних Г. Менделем на основі результатів його експериментів зі схрещування насіння садового гороху. Надалі ці закономірності стали називати законами спадковості і поширили на людину.
закони спадковості
У сучасній редакції закони спадковості формулюються наступним чином.
перший закон- закон домінування (або однаковості) ознаки у нащадків першого покоління. У першому поколінні у нащадка проявляється дію домінантного гена (домінантний
ознака), але не проявляється дію рецесивного гена (рецесивний ознака). У наступних поколіннях у нащадка проявляється дію як домінантного, так і рецесивного гена.
другий закон- закон розщеплення генів у нащадка або закон «чистоти гамет». У нащадка відбувається альтернативне розщеплення (розподіл) генів у гаметах: одна половина гамет несе домінантні гени (А), а інша половина - рецесивні гени (а). Обидва типи генів присутні в соматичних клітинах, не змішуючись і не замінюючи один одного.
третій закон- закон незалежного успадкування неалельних генів або випадкових поєднань спадкових задатків у нащадків. Спадкування двох генів (двох пар ознак) називається дигибридном,більше двох генів (двох пар ознак) - полигибридного.Формула такого наслідування відповідає біномінальної ряду: (3 + 1) п, де η - число генів (пар ознак). Для виведення формули використовуються ґрати Р. Пеннета. З її допомогою розраховуються генотипи організмів, їх кількість і залежність від типів гамет, що містять домінантні і рецесивні гени (табл. 3).
Таблиця 3.Решітка Р. Пеннета
Примітка.А, В, А "і В" - домінантні гени; а, b, а "і b" - рецесивні гени; А "А, В" В, А "В і В" А - домінантні гомозиготні організми; а "а, а" Ь, Ь "а, b" b - рецесивні гомозиготні організми; А "а, а" Ь, а "А, а" В, В "а, В" Ь, Ь "а і Ь" В - домінантні гетерозиготні організми.
Крім трьох законів спадковості, успадкування генів і ознак визначають основні положення хромосомної теорії спадковості.
Хромосомна теорія спадковості
У другому десятилітті XX в. Томас Хент Морган (1866-1945) і його учні (К. Бріджес, Г. Меллер і А. Стертевант) сформулювали основні положення хромосомної теорії спадковості(Усе
Хромосомна теорія спадковості в сучасній редакції включає наступні положення.
Гени розташовані в хромосомах; число генів у хромосомі пропорційно довжині хромосоми. Надалі виявилося: різні гени різні за довжиною, тому не завжди дотримується зазначена пропорція. Наприклад, на хромосомах 5 і 9 генів ідентифіковано більше, ніж на найдовших хромосомах 1 і 2.
Гени розташовані по довжині хромосоми в лінійному порядку. Надалі виділили мобільні генетичні елементи або «стрибають гени» (транспозони), які переміщаються по геному з однієї хромосоми на іншу, порушуючи лінійний порядок розташування.
Алельних гени займають ідентичні локуси гомологічних хромосом. З двох гомологічних хромосом за походженням одна - батьківська, інша - материнська. Їх локуси ідентичні один одному. У них знаходяться алелі одного і того ж гена або алельних гени. Кожен з двох алелей одного і того ж гена є батьківську і материнську копії; в нормі це, як правило, діаллельная модель організаційної структури гена (див. рис. 12). У разі спадкової патології у індивіда може бути тільки один генний локус, і відповідно в ньому буде перебувати один аллель (або батьківський, або материнський); це стан моноаллельності з даного аллелю відзначається, наприклад, при синдромі Шерешевського-Тернера (каріотип: 45, ХО). Інший патологічний варіант - наявність у індивіда одночасно трьох алелей і більш; це стан поліаллельності з даного аллелю - наприклад, три ідентичних алелі одного і того ж гена трьох хромосом 21 при синдромі Дауна (каріотип: 47, ХХ, + 21); три алелі трьох хромосом 13 при синдромі Патау (каріотип: 47, XY + 13) або наявність в каріотипі від 4 до 11 Х-хромосом (4-11 алелей одного і того ж гена - 49, ХХХХ).
Гени однієї хромосоми утворюють групу зчеплення, забезпечуючи спільне успадкування контрольованих ними ознак. Слід зазначити: зчеплення генів з хромосомою постійно порушується в ході кросинговеру - процесу гомологичной рекомбінації або обміну однаковими ділянками (генами і їх фраг-
ментами) між гомологічними хромосомами в першому поділі мейозу. Частота кросинговеру прямо пропорційна відстані між генами. Взаємний обмін алелями батьківського і материнського походження відбувається між усіма парами гомологічних хромосом, за винятком Х-і Y-хромосом. Відкриття кросинговеру також належить школі Т.Х. Моргана. Тепер розглянемо варіанти і типи успадкування генів і ознак.
ВАРІАНТИ І ТИПИ УСПАДКУВАННЯ ГЕНІВ ТА ОЗНАК
В даний час виділяють три варіанти успадкування генів і ознак: моногенний і полігенні при традиційному (класичному) спадкуванні та варіант некласичного або нетрадиційного спадкування.
Моногенне спадкування засноване на першому і другому законах спадковості. Воно має на увазі спадкування одного гена (однієї пари ознак) і відноситься до алельним генам.
Полигенное успадкування засноване на третьому законі спадковості. Воно має на увазі успадкування двох генів (пар ознак) і більш і відноситься до неалельні генам.
Нетрадиційне спадкування є спадкування генів і ознак, що виходить за рамки моногенного і полигенного варіантів.
Моногенне спадкування
Моногенне спадкування нерідко називають простим менделевским спадкуванням.
На основі уявлень про діаллельной моделі структури гена успадкування механізмів взаємодії між батьківським і материнським геномами розглядається окремо для кожної аллельной і неалельних пари.
Типи моногенного успадкування
В рамках моногенного успадкування виділяють:
Аутосомно-домінантний тип (на одній з двох аутосом розташований домінантний ген);
Аутосомно-рецесивний тип (на одній з двох аутосом розташований рецесивний ген);
Х-зчеплений домінантний тип (на Х-хромосомі розташований домінантний ген);
Х-зчеплений рецесивний тип (на Х-хромосомі розташований рецесивний ген);
Y-зчеплений тип або голандріческое спадкування (ген розташований на Y-хромосомі).
Згідно каталогу В. Маккьюсика «Спадкування менделевских ознак у людини» (Інтернет-версія: online - http: www.ncbi.nlm. Nih.gov/Omim), ідентифіковано понад 12,5 тис. Таких фенотипів. Серед них близько 12 тис. - фенотип, успадковані (або імовірно успадковані) аутосомно-домінантно або аутосомнорецессівний (в тому числі 9 тис. Фенотипів з встановленим типом успадкування).
В останні роки отримано ряд даних про спадкування, зчепленому з Х- і Y-хромосомами, на яких локалізовані відповідно
300 і 92 гена.
На рис. 20 приведена карта X-хромосоми і ряд зчеплених з її локусами домінантних і рецесивних захворювань.
Як приклади Х-зчеплених домінантних фенотипів слід привести рідко зустрічаються захворювання: вітамін D-резистентний рахіт або гіпофосфатемія (Хр22.2); синдром нетримання пігменту, тип I (Xp11.1) і тип II (Xq28).
Приклади найбільш поширених захворювань, успадкованих по Х-зчепленим рецесивним типом: гемолітична анемія (Xq21.2 або Xq28), гемофілія А (Xq28.2) і В (Xq27.2), миодистрофия Дюшенна-Беккера (Xp21.2), синдроми Леша -Найяна (Xq26.2),
точкова хондродисплазія Конраді-Хюнерманна (Хр22.2), пігментний ретиніт (Xp21.2-21.3; Xp22).
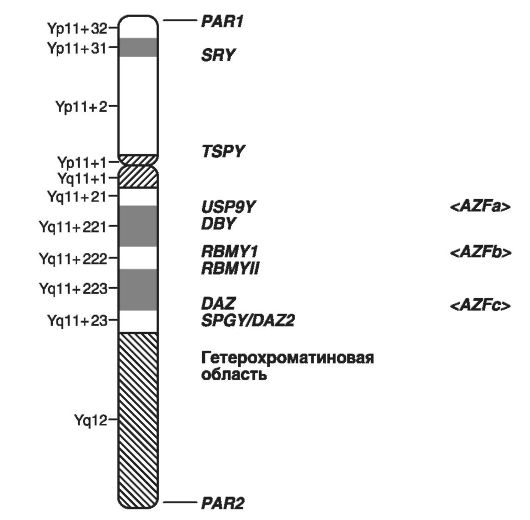
На рис. 21 приведено схематичне зображення локусів Y-хромосоми, в яких розташовані гени, що формують чоловічу стать, і гени, що зумовлюють чоловіче безпліддя.
Як приклади захворювань, зчеплених з Y-хромосомою, слід привести порушення диференціювання статі, форми чоловічого безпліддя у вигляді азооспермії (Yp - фактор 2; Yq11 - фактор 1), гонадобластому і ін. Всього на схемі виділені гени, відповідальні за сім таких захворювань .
Крім того, моногенне спадкування може бути не тільки зчеплене з підлогою (Х-або Y-хромосомою), але і обмежена підлогою.
Мал. 20.Карта X-хромосоми людини (по мультимедійному підручнику «Біологія» Copyright (c), 2007)
Наприклад, ген плешивости проявляється у чоловіків (домінантний ефект) і майже не проявляється у жінок (рецесивний ефект).
Критерії моногенного успадкування
Аутосомно-домінантний тип:
Захворювання регулярно передається з покоління в покоління без пропусків, тобто просліджується в родоводі по вертикалі, крім випадків мутацій de novo (див. розділ 5);
Мал. 21.Схематичне зображення Y-хромосоми (по GenBank, 2003)
Ризик народження хворої дитини, якщо хворий один з батьків, становить 50%;
Здорові індивіди мають здорових нащадків;
У хворого індивіда хворий один з батьків, крім випадків нової мутації;
Обидві статі уражаються з однаковою частотою. Аутосомно-рецесивний тип:
Батьки хворого пробанда (особа, яка звернулася за консультацією до лікаря-генетика) здорові, але аналогічне захворювання виявляється у рідних, двоюрідних і троюрідних сибсов пробанда (його брати і сестри), тобто просліджується в родоводі по горизонталі (в одному поколінні);
У хворого батька народжуються здорові діти;
Ризик народження хворої дитини дорівнює 25% (співвідношення хворих і здорових осіб становить 1: 4);
У разі кровноспоріднених шлюбів між батьками хворого пробанда спостерігається збільшення числа хворих родичів в родоводу.
Х-зчеплений домінантний тип:
У хворого пробанда обов'язково хворий один з батьків;
У хворого батька все дочки хворі, а сини здорові;
У хворої матері народження хворої доньки і хворого сина однаково ймовірно;
У здорових батьків всі діти будуть здорові;
Хворих жінок в 2 рази більше, ніж хворих чоловіків. Х-зчеплений рецесивний тип:
Захворювання спостерігається у чоловіків - родичів хворого пробанда по материнській лінії;
Сини не успадковують захворювання батька;
У хворого батька все дочки здорові і є гетерозиготними носіями гена хвороби батька;
Якщо жінка - гетерозиготний носій гена хвороби, то половина її синів будуть хворі, а всі дочки здорові; причому половина дочок також стануть гетерозиготними носіями гена хвороби.
Критерії голандріческого успадкування поки не розроблені. В останні роки у людини виділено ряд складно успадкованих моногенних і полігенних хвороб:
Дігенние хвороби - рак грудної залози (дві копій гена: гени ВRС1 і ВRС2);
Трігенние хвороби - синдром Барді-Бідля (3 копій гена: гени ВВS1, ВВS2 і ВВS30);
Четирехгенние - пятігенние хвороби: хвороба Альцгеймера (обумовлена чотирма копій гена: PS1, PS2, PS3 і PS4, а також геном пріонів білка, див. Розділ 29);
Полігенні, або мультифакторіальні хвороби (МФБ), обумовлені «генними мережами» - артеріальна гіпертензія (170 генів, включаючи 17 головних генів), бронхіальна астма (близько 20 генів), остеопороз (13 генів), ендометріоз (11 генів) і ін.
полигенное успадкування
Полигенное успадкування нерідко називають мультігенних або мультифакторіальних, маючи на увазі спадкування одночасно не одного, а декількох певних генів, які виявляють своє
дію в специфічних умовах навколишнього середовища, при наявності провокуючих зовнішніх факторів, як правило, підсилюють індивідуальну дію генів, ефект яких підсумовується (адитивна дія).
Критерії полигенного успадкування (всього 5) систематизовані в 1969 р К. Картером, а через 20 років їх доповнили (ще два) Ф. Фогель і А. Мотульський (1989).
Критерії полигенного успадкування
Ризик розвитку мультифакторіальних ознаки (захворювання) визначають наступні чинники.
Успадкованого ознаки або хвороби. Чим вище успадкованого ознаки або захворювання (чим більше успадковано генів, за нього відповідальних), тим вище ризик його розвитку у здорових родичів. наследуемостьознаки (захворювання) - це ступінь впливу на формування даної ознаки (захворювання) спадкових факторів в порівнянні з такою факторів середовища. Наследуемость виражається в абсолютних цифрах (від нуля до одиниці) або відсотках за допомогою коефіцієнта h 2 або К н, який розраховується за формулою: К н = G / Е x 100%, де К н - коефіцієнт успадкування, G - спадкові чинники, Е - фактори навколишнього середовища. У таблиці 4 наведені значення ряду коефіцієнтів успадкування мультифакторіальних ознак і захворювань.
Таблиця 4.Коефіцієнти успадкування мультифакторіальних ознак і захворювань
Ступінь вираженості ознаки або тяжкість перебігу хвороби у пробанда. Чим сильніше виражена ознака або важче протікає
захворювання у хворого родича, тим вище ризик його розвитку у здорових родичів.
Спільність генів у пробанда і його родичів (або близький ступінь споріднення з хворим родичем). Чим більше спільних генів у хворого і його родичів, тим вище ризик розвитку у останніх ознаки або захворювання. Наприклад, популяційна частота псоріазу становить 0,75%. У родичів I ступеня спорідненості частота його розвитку - 5,6%, у родичів II ступеня споріднення - 3,0-3,5%, у родичів III ступеня споріднення - 1,75%, у родичів IV ступеня споріднення - 0,75% .
Рідко битий підлогу. Мультифакторіальний ознака або захворювання проявляється частіше у осіб рідко вражається статі (критерій, названий ефектом Картера).Наприклад, вроджений пілоростеноз у хлопчиків зустрічається в 2-5 разів частіше, ніж у дівчаток, тобто в даному випадку жіноча стать - рідко битий підлогу. Однак частота цієї хвороби у майбутніх дітей уражених пілоростенозом дівчаток досягне 10-20%, тоді як у майбутніх дітей уражених пілоростенозом хлопчиків - тільки 2-6%. Інший приклад - виразкова хвороба шлунка та дванадцятипалої кишки, як правило, виявляється в осіб чоловічої статі і набагато рідше - у осіб жіночої статі. Однак її частота у дітей хворої жінки вище, ніж у дітей хворого чоловіка.
Число хворих родичів. Чим більше в родоводу родичів, що мають мультифакторіальний ознака або захворювання, тим вище ризик його розвитку у нащадків (табл. 5).
Таблиця 5.Ризик розвитку мультифакторіальних ознаки або захворювання у пробанда в залежності від числа його хворих родичів
Загальний ризик для дітей, якщо їхні батьки здорові, становить 5-10%; якщо хворий один з батьків - 10-20%; якщо хворі обоє батьків - до 40%;
додаткові критерії
Близнюковий критерій. Якщо конкордантность (схожість) монозиготних близнюків по якоюсь ознакою або захворювання в 4 рази вище конкордантности у дизиготних близнюків, то ця ознака або захворювання успадковується по полігенною варіанту.
Критерій сегрегаційного відносини уражених сибсов в сім'ях з одним хворим або двома здоровими батьками. Якщо частка хворих сибсов в сім'ях з одним хворим батьком вище в 2,5 рази (і більше), ніж частка хворих сибсов в сім'ях з двома здоровими батьками, то особливо ймовірний полігенний варіант успадкування. Якщо вказане співвідношення менше 2,5, то полигенное успадкування також не виключено.
Однак класичні варіанти успадкування генів і ознак мають місце лише в меншій частині всіх випадків такого наслідування, тоді як майже 2/3 з них - нетрадиційні варіанти.
нетрадиційне спадкування
Нетрадиційне спадкування відзначається при багатьох спадкових хворобах людини. В останні роки до них віднесені раніше погано вивчені або взагалі невідомі захворювання 6 класів: хвороби накопичення (44 нозології; див. Розділ 21); пероксисомні хвороби (17 нозологій; див. розділ 26); мітохондріальні хвороби (виділено понад 200 нозологій, передбачається наявність ще близько 1150; див. розділ 26); хвороби імпринтингу (виділені 24 нозології; див. розділ 28); хвороби експансії числа нуклеотидних повторів (27 нозологій; див. розділ 27); пріонні хвороби (9 нозологій; див. розділ 29).
В цілому на частку хвороб нетрадиційного спадкування припадає 321 нозологічна одиниця (без урахування 1150 мітохондріальних хвороб). При спадкуванні захворювань перших трьох класів йдеться про материнському спадкуванні,тому лізосоми, мітохондрії і пероксисоми, будучи цитоплазматическими структурами соматичних клітин, успадковуються виключно по лінії мати-дочка і ніколи не передаються по лініях мати-син, батько-син або батько-дочка, що обумовлено біологічним матріархатом(Див. Глави 5 і 12).
Разом з тим, при ряді мітохондріальних хвороб, крім чисто материнського успадкування, може спостерігатися змішане з материнським моногенне успадкування. Воно пов'язане з порушеннями взаємодії між мітохондріальних (мтДНК) і ядерним (ДНК) геномами. Зокрема, гени, що кодують мітохондріальні білки, знаходяться або в мітохондріях (гени двох типів рРНК, 22 типів тРНК, 13 типів поліпептидів), або в ядрі клітини (їх приблизно 1 150). Ядерні гени транскрибуються в ядрі, транслюються в цитоплазмі, а результати їх експресії «імпортуються» мітохондріями.
Зазвичай всі копії мтДНК ідентичні між собою (гомоплазія). У випадках, коли в мтДНК виникають мутації (їх частота в 10 разів частіше, ніж в ядерній ДНК), можлива наявність в одній клітці двох типів мтДНК (гетероплазія), і тоді процес поширення мутантною і нормальної мтДНК називається репликативной сегрегацією(Див. Розділ 2).
У разі хвороб імпринтингу або епігеномний виключення з експресії локусів хромосом одного з батьків (див. Вище) фенотипічні прояви дії гена можуть змінюватися в результаті трьох причин: делеция гена (генетичний імпринтинг), однородітельская ізодісомія (хромосомний імпринтинг) і порушення генної експресії в центрі імпринтингу .
генетичний импринтингзаснований на механізмі специфічного метилування цитозинових підстав молекули ДНК, вимикати транскрипцію гена (див. розділ 28).
В даний час передбачається існування близько 200 генів, схильних до такого імпринтингу і мають тканеспеціфіческіе моноаллельную експресію, а також виявлені три кластера генів, локалізованих в критичних областях хромосом (7q32, 11р15 і 15q11.2-q13) і надають прямий вплив на розвиток спадкової патології по таким механізмом, в тому числі пухлин.
геномної імпрінтінгзаснований на однородітельской ізодісоміі по конкретній хромосомі або материнського, або батьківського походження. Він спостерігається при ряді спадкових і вроджених захворювань, таких, як: синдроми Ангельмана (причина - однородітельская ізодісомія по батьківській хромосомі 15) і Прадера-Віллі (ізодісомія по материнській хромосомі 15); транзиторний неонатальний цукровий діабет (батьківська хромосома 6); синдром Сільвера-Рассела (материнська хромосома 7); синдром
Беквітта-Видемана (батьківська хромосома 11); затримка фізичного і моторного розвитку, гіпотонія і передчасний статевий розвиток (материнська хромосома 14); затримка внутрішньоутробного розвитку (ЗВУР) плода (материнська хромосома 7), а також ЗВУР з обмеженим плацентарних мозаїцизмом (материнська хромосома 16).
Ефект геномного імпринтингу має місце також при міхурово заметі. Відзначено 4 типи порушень:
Андрогенез або подвійний набір хромосом (2n): два сперматозоїда з Х-хромосомами і яйцеклітина без ядра;
Гіногенез або подвійний набір хромосом (2n): яйцеклітина з подвійним набором хромосом, сперматозоїди не беруть участі в заплідненні;
Андроїд або потрійний набір хромосом (3n): 2 батьківських і 1 материнська;
Гіноід або потрійний набір хромосом (3n): 2 материнські і 1 батьківська.
Імпрінтінг в результаті помилок генної експресії в центрі імпринтингу
відбувається також при синдромах Ангельмана і Прадера-Віллі. Він обумовлений тим, що обоє батьків передають хворому нащадку гени, що несуть їх специфічні властивості, тобто гени батька і матері активовані або супрессіровани у нащадка по-різному (так звані імпринтовані гени).Причому по лінії як матері, так і батька успадковуються саме батьківські порушення генної експресії в центрі імпринтингу.
В даний час виділено не менше 60 імпринтованих генів (або їх транскриптів) на 1, 5-7, 11, 13, 15, 19 і 20 ваутосомах і Х-хромосомі.
Слід зазначити, що при синдромах Ангельмана і Прадера- Віллі також виявлені близько розташовані, але протилежно імпринтовані гени. їх назвали генами-кандидатами захворювань(В сім'ях з повторними випадками цих синдромів). Причому геникандідати синдрому Ангельмана експресуватися виключно на материнській хромосомі, але репресували на батьківській хромосомі, тоді як геникандідати синдрому Прадера-Віллі експресуватися на батьківській хромосомі, але репресували на материнській хромосомі (див. Розділ 28).
Таким чином, імпринтинг є результат якісних, а не кількісних змін спадкового матеріалу.
В кінці XX в. виділили також раніше невідомий клас хвороб з нетрадиційним спадкуванням - хвороб експансії нуклео-
тідних повторів,пов'язаних зі збільшенням числа кодують і не кодують послідовностей нуклеотидів в результаті динамічних мутацій. Динамічна мутація «рухається» від стану фенотипически не проявляє премутаціі до стану фенотипически виявляється повною мутації. Такі мутації лежать в основі ряду тяжких спадкових нейродегенеративних захворювань (27 нозологій; див. Розділ 27). Наприклад, повтор ЦЦГ характерний для синдрому Мартіна-Белл (Xq23), повтор ГГц - для другого варіанту цього синдрому (Xq28); повтор ЦТГ - для спінобульбарной м'язової атрофії (Xq11-12); ЦАГ - для миотонической дистрофії (19q13.3); ЦТГ - для хореї Гентингтона (4р16.3); ЦТГ - для спіномозжечковий атаксії, тип I (6р21.3); ЦТГ - для хвороби Мачадо-Джозефа (14q32.1); ТТЦ - для атаксії Фридрейха (9р13). У всіх випадках відбувається поступове накопичення (експансія) критичного числа нуклеотидних повторів.
Паралельно з експансією в кожному наступному поколінні наростає тяжкість перебігу захворювання - антиципация.
Показники Взаємодія генів ЯК ДОДАТКОВІ ПОНЯТТЯ ГЕНОМІКИ та протеоміки
Згідно закономірностям генетики, батьківський і материнський геноми, які об'єдналися в генотипі однієї клітини і цілого організму, вступають між собою у взаємодію протягом усього онтогенезу, створюючи індивідуальну зовнішню і внутрішню характеристику, фенотип організму.
Виділяють три групи механізмів взаємодії між батьківськими генами і відповідно три групи показників, що відносяться до додаткових понять геноміки і протеоміки. Це взаємодії між:
Аллельними генами; їх характеризують: домінування, рецессірованіе, кодоминирование, неповне і умовне домінування, наддомінування;
Неалельних генами; їх характеризують: епистаз, компліментарність, полімерія;
Окремим геном і генотипом (як системою генів); їх характеризують: експресивність, пенетрантність, ефект положення,
генокопірованіе, ефект плейотропії, алельних серії і фенокопірованіе.
Розглянемо ці механізми як додаткові поняття геноміки і протеоміки.
Механізми взаємодії між аллельними генами
Домінування і рецессірованіе
Якщо функціональний стан одного алеля (наприклад, материнський аллель) не залежить від стану іншого аллеля (батьківський аллель), то у нащадка проявиться ознака, контрольований материнським аллелем. Такий ген і контрольований ним ознака називаються домінатний.
Перший опис ефекту домінування відноситься до 1905 року, коли в родоводі сім'ї хворого з брахідактилією була відзначена короткопалость. Іншими прикладами служать: білий локон, «куряча сліпота», габсбургська губа, полідактилія (багатопалості), синдактилія (зрощення м'яких або кісткових тканин фаланг), арахнодактилія ( «павукові пальці»), хондродистрофия, а також численні форми аутосомно-домінантних захворювань, наприклад хорея Гентингтона (4р16.3).
Якщо функціональний стан одного алеля (наприклад, материнський аллель) залежить від стану іншого аллеля (батьківський аллель), то у нащадка проявиться ознака, контрольований і материнським, і батьківським алелями одночасно. При цьому і сам ген, і контрольований ним ознака називаються рецесивними.
Приклади успадкування рецесивних ознак і фенотипів: альбінізм, м'які пряме волосся, кирпатий ніс, світлі очі, резусотріцательная I група крові, нездатність відчувати смак фенілтіокарбаміда; численні форми аутосомно-рецесивних захворювань, наприклад копій гена фенілкетонурії (12q24.2; 4р15.1).
У деяких випадках домінантність і рецессивность генів погано співвідносяться з домінантністю і рецесивних ознак. Наприклад, епікант у монголоїдів контролюється домінантним геном, а у бушменів і готтентотів - рецесивним геном.
Інший приклад - згадані вище ген і ознака плешивости, які проявляються у чоловіків як домінантні, а у жінок як рецесивні. Як виявилося, такий механізм обумовлений дією гормонів, тобто має місце залежне від статі (контрольоване підлогою) успадкування.
неповне домінування
Про неповному домінуванні,або проміжному дії генів (прояві ознак), говорять при ослабленні дії домінантного гена в присутності рецесивного гена, тобто у гетерозигот. Однак чітку межу між проміжним дією і домінантністю з одного боку, а також проміжним дією і рецесивних з іншого боку, провести не можна. Наприклад, пігментація шкіри у людини варіює від білого кольору у альбіносів до чорного кольору у негрів. Від шлюбів між білими і неграми народжуються мулати, що мають проміжний колір шкіри.
Інший приклад неповного домінування - відмінності по 6 типів співочого голоси, які контролюються однією аллельной парою. Зокрема, баритон і меццо-сопрано спостерігаються тільки у гетерозигот, тоді як тенор і бас, альт і сопрано характерні для гомозигот.
При подальших дослідженнях, правда, виявилося: пігментація шкіри і тип співочого голосу визначаються не тільки цим механізмом взаємодії, але і незалежними один від одного факторами: впливом статевих гормонів, ефектом полімерії, зчепленим з підлогою або залежним від статі спадкуванням.
Умовне (нестійке) домінування
При нестійкому або умовному, домінуванніпрояв ознаки у гетерозигот залежить від генотипу і зовнішніх умов (модифікуючу вплив генотипу на головний ген, пенетрантность гена, місце розташування гена в складі хромосоми, вплив температури).
кодомінування
Якщо алельних гени активні в однаковій мірі (мають однаковий домінантним дією), то це кодоминирование.Класичний його приклад - успадкування IV групи крові (за системою АВО), яка визначається трьома алелями, розташованими в 9-й хромосомі (множинність алелей). Серед них - два домінантних алелі (I A і! В) і один рецесивний аллель (I 0). Попарне поєднання цих алелей дає 4 групи крові:
- перша група- наявність двох однакових рецесивних алелей - I 0 I 0 (гомозигота), що обумовлюють присутність в сироватці крові альфа- і бета-антитіл;
- друга група- наявність двох однакових домінантних алелів I A I A (гомозигота) або двох різних алелей I A I 0 (гетерозигота), що обумовлюють присутність в сироватці крові бета-антитіл;
- третя група- наявність двох однакових домінантних алелів М В (гомозигота) або двох різних алелей М 0 (гетерозигота); в сироватці крові присутні альфа-антитіла;
- четверта група- наявність двох різних домінантних алелів! А! В (гетерозигота); в сироватці крові немає антитіл, обидва алелі взаємодіють з однаковою силою, нейтралізуючи одне одного.
Ще один приклад Кодомінантність - успадкування серповидноклітинної анемії, яка є аутосомно-рецесивним захворюванням (11р15). В даному випадку спостерігається гомозиготность (2 патологічних алелі одного гена, контролюючого синтез дефектного гемоглобіну). Такі гомозиготи мають характерну симптоматику, але вони несприйнятливі до малярії, бо малярійний плазмодій не чути на дефектному гемоглобіні.
Разом з тим, в гетерозиготному організмі одночасно присутні нормальний і дефектний аллели одного і того ж гена. Причому обидва алелі дають однаковий домінантний ефект, і тому в клітинах одночасно синтезуються два види гемоглобіну (нормальний і аномальний). У таких гетерозиготних носіїв патологічного гена симптомів серповидноклітинної анемії немає або вона проявляється в легкій формі і тільки в умовах кисневої недостатності.
Наддомінування
У ряді випадків аллели, що знаходяться в гетерозиготному стані, фенотипически проявляються сильніше, ніж аллели, що знаходяться в гомозиготному стані (Ефект сверхдоминирования).Таке їх прояв нагадує ефект гетерозисуу рослин (гібридна потужність або сила). Так, в разі шлюбів між представниками різних рас показники здоров'я їх нащадків перевершують таке самих батьків: діти відрізняються більш високими життєздатністю, тривалістю життя і ін.
Механізми взаємодії між неалельних генами
епістаз
епістаз- придушення дії гена, що знаходиться в одній неалельні парі, дією гена з іншої неалельні пари, наприклад придушення геном А гена В, тобто A\u003e B або A\u003e bb. Виділяють домінантний і рецесивний епістаз.
Домінантний епістаз:домінантний аллель однієї неалельні пари, що знаходиться в гомозиготному (АА) або гетерозиготному (Аа) стані, пригнічує прояв неалельні до нього домінантного алеля іншого алельних пари, що знаходиться в стані АА або Аа. Гени, що дають домінантний ефект, називаються епістатичний генамиабо супрессорами (інгібіторами). 0ни можуть бути як домінантними, так і рецесивними. Придушуються гени називаються гіпостатичними генами.
Якщо гени, що знаходяться в інших неалельних парах, підсилюють домінантне дію Епістатичний генів, то вони називаються генами-модифікаторами(Интенсификаторами).
Такий тип взаємодії характерний для неалельних генів, що беруть участь в регуляції онтогенезу, наприклад генів імунної відповіді (генна мережа - 2190 генів; див. Розділ 15) або генів еритропоезу (генна мережа - 200 генів).
Можливі два варіанти домінантного епістазу:
Гомозиготи з рецесивними алелями (аа) відрізняються за фенотипом від гомозигот з домінантними алелями (АА);
Гомозиготи по домінантним аллелям (АА) не відрізняються за фенотипом від гомозигот по рецесивним аллелям (аа).
рецесивний епістазпроявляється в тому, що рецесивний аллель одного гена пригнічує дію неалельні йому домінантного гена (аа\u003e В), а між домінантними генами спостерігається компліментарність (див. нижче). Прикладом рецесивного епістазу у людини служить «бомбейський феномен», пов'язаний з народженням дітей з I (I 0 I 0) і IV (I А I В) групами крові від батьків з I (I 0 I 0) і II (IAI °) групами крові, тоді як теоретично від таких батьків повинні народжуватися діти з I (I 0 I 0) або II (IAI 0) групами крові. Феномен можна пояснити або наявністю не розпізнає у одного з батьків рідкісного гетерозиготного варіанту III групи крові (IBI 0), або наявністю в генотипі дитини з IV групою крові (I А I В) рецесивних генів-модифікаторів, які в гомозиготному стані пригнічують експресію антигенів, знаходяться на поверхні еритроцитів, тобто дають непередбачуваний фенотипический ефект.
Крім рецесивного епістазу, виділений подвійний рецесивний епістаз;при ньому у рецесивних генів власне фенотипічніпрояв, а в подвійних Гомозигота рецесивні аллели пригнічують один одного: аа\u003e bb, bb\u003e аа.
комплементарність
комплементарність- тип взаємодії не менше ніж двох домінантних неалельних генів з декількох пар з різним поєднанням домінантних і рецесивних алелей, що обумовлюють розвиток нової ознаки, відмінного від батьківських варіантів.
Відомо три типи комплементарності:
Домінантні аллели (АВ) розрізняються по фенотипическому прояву;
Домінантні аллели (АВ) подібні за фенотипическому прояву;
У домінантних (А) і рецесивних (а) алелей з декількох неалельних пар - самостійне фенотипічніпрояв.
Наприклад, у людини нормальний слух обумовлений взаємодією декількох пар неалельних генів, але в парах повинен знаходитися як мінімум один домінантний аллель. Якщо ж людина виявиться рецессивной гомозиготой (хоча б по одній парі неалельних генів), то він буде глухим.
Іншими прикладами комплементарності служать фенотип онкологічних хворих з ретинобластому і нефробластома (див. Розділ 25).
Полімерія і формування кількісних ознак
полімерія- це обумовленість ознаки або фенотипу взаємодією генів, локалізованих в декількох неалельних парах і дають однаковий ефект. Такі гени називаються полімерними генамиабо полігенами. Ступінь прояву ознаки (фенотипу) залежить як від числа домінантних генів в неалельних парах, так і від числа неалельних пар.
Такі ознаки називаються кількісними ознаками.0ни істотно відрізняються від якісних ознак.
Якщо кількісні ознаки контролюються генами, успадкованими полигенно, і проявляються в багатьох станах як перехідні форми, то якісні ознакиконтролюються генами, успадкованими моногенно, і проявляються тільки в альтернативних станах без перехідних форм.
Прикладами кількісних ознак є: згадувані вище варьирующая пігментація шкіри у людини і наявність проміжного кольору шкіри у мулатів.
Інші приклади кількісних ознак: рівень (стан) здоров'я людини, тривалість життя, інтелектуальні здібності, маса і довжина тіла.
В останні роки виділено феномен взаємодії між численними неалельних генами - кумулятивна полімерія.В даному випадку мова йде про аддитивном (суммирующем) дії генів, кожен з яких робить свій (часто невелике) вплив на ознаку. Саме кумулятивна полімерія формує згадані вище генні мережі, контролюючі значну частину кількісних ознак організму.
Серед генів, що впливають на кількісний ознака, можуть виявитися один головний ген і ряд більш слабких у порівнянні з ним генів (поліг). Дія головного гена часом значно перевершує таке інших генів, і контрольований головним геном ознака успадковується як менделевским (моногенний варіант успадкування), а ознаки, контрольовані полігенами, успадковуються по полігенною варіанту. Приклад - успадкування карликовості, обумовленої головним геном в разі ахондроплазії, тоді як в нормальній популяції зростання людини визначається адитивну дію полигенов.
МЕХАНІЗМИ Взаємодія між
ОКРЕМИХ ГЕНОМ І генотипу
Експресивність і пенетрантність
Ці поняття вперше введені в 1926 р Н.В. ТімофеевимРессовскім і 0. Фогтом для опису варьирующего прояву ознак і контролюючих їх генів. експресивністьє ступінь вираженості (варіювання) одного і того ж ознаки у різних осіб, що мають ген, що контролює цей показник. Спостерігається низька і висока експресивність. Розглянемо, наприклад, різну вираженість риніту (нежиті) у трьох різних хворих (А, Б і С) з одним і тим же діагнозом 0РВІ. У хворого А риніт виражений в легкого ступеня ( «шмигання носом»), що дозволяє протягом дня обходитися одним носовою хусткою; у хворого Б риніт виражений у стані середнього ступеню (щодня 2-3 носових хустки); у хворого С - висока ступінь вираженості риніту (5-6 носових хусток). Коли говорять про експресивності не окремо взятого ознаки, а захворювання в цілому, лікарі часто оцінюють стан хворого як задовільний або середнього ступеня тяжкості, або як важкий,
тобто в даному випадку поняття експресивності аналогічно поняттю «тяжкість перебігу хвороби».
пенетрантность- це ймовірність прояву одного і того ж ознаки у різних осіб, що мають ген, що контролює цей показник. Пенетрантность вимірюється в відсотку осіб з певною ознакою від загального числа осіб, які є носіями гена, контролюючого дана ознака. 0на буває неповної або повної.
Прикладом захворювання з неповною пенетрантностью служить все той же риніт при 0РВІ. Так, можна вважати, що у хворого А немає риніту (але є інші ознаки захворювання), тоді як у хворих В і С риніт є. Тому в даному випадку пенетрантность риніту становить 66,6%.
Приклад захворювання з повною пенетрантностью - аутосомнодомінантному хорея Гентингтона(4р16). 0на маніфестує переважно у осіб у віці 31-55 років (77% випадків), у інших же хворих - в іншому віці: як в перші роки життя, так і в 65, 75 років і більше. Важливо підкреслити: якщо ген цієї хвороби переданий нащадку від одного з батьків, то хвороба проявиться обов'язково, в чому полягає повна пенетрантність. Правда, пацієнт не завжди доживає до маніфестації хореї Гентингтона, вмираючи від іншої причини.
ефект положення
Інший тип залежності дії гена від генотипу - ефект положення.0ткрил його А. Стертевант (1925). Суть ефекту - в зміні експресії гена при зміні займаного нею положення (позиції) в хромосомі (в ряду нуклеотиднихпослідовностей).
За сучасними уявленнями, ефект положення не пов'язаний з порушенням структури гена: він і його промоторних область зберігаються як одиниця транскрипції. Отже, ефект положення - епігеномний подія, яке визначається трьома умовами:
У промоторі відбувається ініціація транскрипції;
Регуляторні елементи (енхансери і сайленсери), що містять сайти зв'язування факторів транскрипції, збільшують специфічність транскрипційного комплексу на промоторі;
Організація хроматину в районі локусу сприяє підвищеній чутливості до дії нуклеаз.
Рівень експресії залежить від місця розташування гена в геномі: або в районах конденсованого гетерохроматину, або в районах
недеконденсірованного хроматину (еухроматину), який деконденсірован в інтерфазі, містить більшість генів і реплицируется на початку S-фази. У свою чергу, гетерохроматин конденсованих протягом всього клітинного циклу, реплицируется в кінці S-фази і містить в основному повторювані послідовності. Центромерного області хромосом складаються з структурного гетерохроматину або щільно конденсованого хроматину, що містить повторюють послідовності.
У зв'язку з цими особливостями при формуванні структурних перебудов хромосом їх розриви ведуть до зміни положення генів, що супроводжується зміною їх експресії (рис. 22). Домінантний ген А, який опинився поблизу місця розриву хромосоми, в більшій мірі втрачає свій вплив, ніж ген В, розташований далі від місця розриву. Іншими словами, ослаблення ефекту домінантного гена пропорційно відстані між ним і точкою розриву хромосоми.
В інших випадках хромосомная перебудова може:
Відокремити Транскрипційні одиницю від регуляторного району, повністю нейтралізувавши його вплив на ген (відсутність енхансера знижує або усуває транскрипцію у відповідній тканини); в свою чергу, поділ гена і його сайленсери призведе до аномального збільшення експресії;
Перемістити ген в район енхансера іншого гена, що викликає неадекватну експресію (наприклад, при лімфомі Беркітта транслокация переміщує ген c-myc під контроль енхансера іммуноглобіна);
Перемістити ген і його регуляторні послідовності в район іншого гена, що знижує рівень експресії першого гена;
Викликати мозаїчний ефект положення (наприклад, червоний і білий колір очей у дрозофіли).
 Мал. 22.Ефект положення в разі розриву хромосоми:
Мал. 22.Ефект положення в разі розриву хромосоми:
З - центромера; р і q - коротке і довге плечі хромосоми; Х - генний
локус; А і В - домінантні гени
Інший приклад прояву ефекту положення - індивіди з групами крові за системою Rh. Зокрема, особи з генним комплексом CDE / cDe мають ті ж гени, що і особи з генним комплексом cDe / CDE (де велика літера - домінантний ген, а прописна буква - рецесивний ген). Однак у перших багато антигену Е і мало антигену С, а у других - навпаки, що пояснюється неоднаковою локалізацією генів С і Е - на одній хромосомі або на різних хромосомах відповідно.
Ще один приклад - аутосомно-домінантна особі-лопаточноплечевая миодистрофия типу 1А, обумовлена делецией вариабельного ділянки в субтеломерной частини довгого плеча хромосоми (розташований в сегменті 4q35), який не містить генів, але має нуклеотидні послідовності розмірами 20-250 кб. Делеция цих послідовностей призводить до зміни структури гетерохроматина в даній області хромосоми і опосередкованого пригнічення транскрипції довколишнього гена (FRG1), розташованого проксимально від точки розриву хромосоми.
Назвемо ще ряд спадкових захворювань, пов'язаних з ефектом положення. Серед них:
Анірідія в результаті делеції гена, розташованого в локусі
Голопрозенцефалія - мутація в гені SSN (7q36);
Краніосиностоз Сатре-Котц - мутація в гені TWIST (7p21);
Цефалополісіндактілія Грейга в результаті мутації генаСLIЗ
Синдром Рігер - ген PITX2 (4q25-q27);
XY-реверсія статі - мутація в гені SRY (Xq).
Генокопірованіе і його причини
Один і той самий ознака (група ознак) буває обумовлений різними генетичними причинами (або гетерогенність). Такий ефект, за пропозицією німецького генетика Х. Нахтхайма, отримав в середині 40-х років XX ст. назва генокопірованія.Відомі три групи причин генокопірованія.
Причини першої групиоб'єднує гетерогенність за рахунок полілокусності, або дії різних генів, розташованих в різних локусах на різних хромосомах. Наприклад, серед спадкових хвороб обміну складних цукрів - глюкозоаміногліканов виділені 19 типів (підтипів) мукополисахаридозов. Всі типи харак-
теризують дефектами різних ферментів, але проявляються однієї і тієї ж (або подібною) симптоматикою гаргоіліческого дісморфізмаабо фенотипу дзвонаря Квазімодо - головного героя роману «Собор Паризької Богоматері» класика французької літератури Віктора Гюго. Схожий фенотип нерідко спостерігається і при муколіпідозов (порушеннях обміну ліпідів).
Інший приклад полілокусності - фенілкетонурія. Зараз виділені не тільки її класичний тип, обумовлений дефіцитом фенілаланін-4-гідроксилази (12q24.2), але і три атипові форми: одна викликана дефіцитом дігідроптерідінредуктази (4р15.1), а ще дві - дефіцитом ферментів пірувоілтетрагідроптерін-синтетази і тетрагідробіоптеріна (відповідні гени поки не визначені).
Додаткові приклади полілокусності: глікогенози (10 копій гена), синдром Еллерс-Данлоса (8), нейрофібраматоз Реклингаузена (6), вроджений гіпотиреоз (5), гемолітична анемія (5), хвороба Альцгеймера (5), синдром Барді-Бідля (3), рак грудної залози (2).
Причини другої групиоб'єднує внутрілокусная гетерогенність. Вона обумовлена або множинним алелізм (див. Розділ 2), або наявністю генетичних компаундов,або подвійних гетерозигот, що мають два однакових патологічних аллеля в ідентичних локусах гомологічних хромосом. Приклад останнього - гетерозиготна бета-таласемія (11р15.5), що формується в результаті делеций двох генів, що кодують бета-ланцюга глобинов, що веде до підвищеного вмісту гемоглобіну HbA 2 і підвищеного (або нормальному) рівнем гемоглобіну HbF.
Причини третьої групиоб'єднує гетерогенність за рахунок мутацій в різних точках одного й того ж гена. Приклад - муковісцидоз (7q31-q32), що розвивається через наявність майже 1000 точкових мутацій в гені, відповідальному за хвороба. При загальній довжині гена муковісцидозу (250 тис. Н.п.) в ньому передбачається виявити до 5000 таких мутацій. Даний ген кодує білок, відповідальний за трансмембранний перенос іонів хлору, що веде до збільшення в'язкості секрету екзокринних залоз (потових, слинних, під'язикові і ін.) І закупорці їх проток.
Інший приклад - класична фенілкетонурія, зумовлена наявністю 50 точкових мутацій в гені, що кодує фенілаланін-4-гідроксилази (12q24.2); всього при цій хворобі передбачається виявити більше 500 точкових мутацій гена. Більшість їх виникає
через поліморфізму по довжині рестрикційних фрагментів (RFLP) або по числу тандемних повторів (VNTP). Встановлено: головна мутація гена фенілкетонурії в слов'янських популяціях - R408 W /
ефект плейотропії
Вищезазначена неоднозначність характеру зв'язків між генами і ознаками виражається також в ефекті плейотропіїабо плейотропних дії, коли один ген викликає формування цілого ряду ознак.
Наприклад, ген аутосомно-рецесивною атаксії-телеангіектазії, або синдрому Луї-Бар(11q23.2) відповідальний за одночасне ураження не менше шести систем організму (нервова і імунна системи, шкірні покриви, слизові оболонки органів дихання і шлунково-кишкового тракту, а також кон'юнктива очей).
Інші приклади: ген синдрому Барді-Бідля(16q21) обумовлює слабоумство, полідактилія, ожиріння, пігментну дегенерацію сітківки; ген анемії Фанконі (20q13.2-13.3), який контролює активність топоізомерази I, викликає анемію, тромбоцитопенію, лейкопенію, мікроцефалія, аплазию променевої кістки, гіпоплазію п'ясткової кістки I пальця, пороки розвитку серця і нирок, гипоспадию, пігментні плями шкіри, підвищену ламкість хромосом .
Виділяють первинну і вторинну плейотропії. первинна плейотропіяобумовлена біохімічними механізмами дії мутантного білка-ферменту (наприклад, недостатністю фенілаланін-4-гідроксилази при фенілкетонурії).
вторинна плейотропіяобумовлена ускладненнями патологічного процесу, що розвинувся в результаті первинної плейотропії. Наприклад, за рахунок посиленого кровотворення і гемосидероза паренхіматозних органів у хворого з таласемією виникають потовщення кісток черепа і гепатоліенальнийсиндром.
алельні серії
Точкові мутації одного гена можуть зумовити розвиток не тільки одного, але і різних симптомокомплексів. У другому випадку грають роль алельних серії одного і того ж гена.Наприклад, мутації гена адренорецептора, зчепленого з Х-хромосомою, стають причиною хвороби Кеннеді,якщо вони захоплюють область тринуклеотидних повторів в екзоні 1, але здатні привести до син-
дром Моррісаабо тестикулярной фемінізації (фенотипически - дівчинка, генотипически - хлопчик, тобто наявність жіночого фенотипу при чоловічому каріотипі), якщо захоплюють інші нуклеотидні послідовності даного гена (мутація в андрогенсвязивающем домені).
Іншим прикладом гетерогенності за типом аллельной серії служать точкові мутації гена рецептора тирозинкінази - RET, що викликають чотири спадкові хвороби: сімейну мозкову карциному щитовидної залози, хвороба Гіршпрунга, множинну ендокринну неоплазіїдвох типів - 2А (МЕН2А) і 2В (МЕН2В).
Хоча описуваний феномен відкритий відносно недавно, вже відомо понад 100 моногенних хвороб, обумовлених мутаціями одного гена за типом аллельной серії. Їх причинами служать:
Різні послідовності нуклеотидів (різні точки гена), контролюючі функціонально різні домени білка;
Наявність в одному і тому ж гені модифікуючий мутантного аллеля (алельних поліморфізм);
Вплив генетичного оточення на прояв мутантного аллеля (в тому числі взаємодія мутантного аллеля з алелями одного або декількох генів-модифікаторів).
Фенокопірованіе
Фенокопірованіе- це копіювання симптомокомплексу ненаследственной хвороби (що сформувалася під впливом чинників середовища) як симптомокомплексу спадкової генетичної хвороби.
Наприклад, симптоматика ендемічного зобу відповідає симптоматиці вродженого гіпотиреозу, але в першому випадку причина - дефіцит (або повна відсутність) неорганічного йоду в питній воді та продуктах харчування (тобто має місце фенокопірованіе), а в другому - різні генетичні чинники (т. е. має місце генокопірованіе).
Інший приклад генокопірованія і фенокопірованія симптоматики - сліпота. Відомо, що зір контролюється групою генів (генна мережа), що взаємодіють один з одним в ході онтогенезу і забезпечують розвиток і підтримку функціонування очей і головного мозку. Різні причини порушення цілісності цієї системи здатні зумовити один і той же результат (сліпоту).
Так, внаслідок генокопірованія сліпота може розвинутися через дефекти кришталика, що визначаються одними генами, дефектів сітківки, що визначаються іншими генами, і дефектів рогівки, що визначаються третіми генами.
Внаслідок же фенокопірованія за рахунок екзогенного впливу середовища сліпота може розвинутися через недотримання нормативів освітленості житлових і виробничих приміщень або в результаті очного травматизму.
Просте копіювання схем вдалого розведення не принесе очікуваного успіху собаківникові. Іноді незнання разведенца якості предків його племінних собак приносить породі так багато шкоди, що на той час, коли він усвідомлює масштаби збитку, часто вже занадто пізно, щоб що-небудь виправити. Тому собаківник, перш ніж зайнятися розведенням собак, зобов'язаний дізнатися якомога більше про обраної породі, про недоліки, найбільш часто зустрічаються в ній, про кращих і гірших предків племінних собак, з якими він збирається вести
розведення.
Собаківники повинні весь час пам'ятати, що вони ТІЛЬКИ Тимчасові ОПІКУНИ обраної ними породи!
Найчастіше причиною недоліків і відхилень від норми є рецесивні гени. З приблизною достовірністю можна вважати: якщо нащадок проявляє характеристику, якої не було ні в одного з батьків, то ця характеристика визначається рецесивним геном.
Наприклад, якщо у обох батьків темні очі або мочки носа, а у цуценяти вони світлі, значить, у обох батьків був предок з рецесивним геном, визначальним така ознака, і вони цей ген успадкували. При зведенні цих собак в пару гени зійшлися разом - в результаті темноокі батьки дали ясноокого нащадка. Якщо цього нащадка спарити з аналогічною собакою зі світлими очима, що має темнооких батьків, жоден з них цуценят не матиме темних очей.
Цей принцип можна застосувати також до пороків, які визначаються простим рецесивним геном, як, наприклад, "вовча паща" або глухота. Звідси зрозуміло як легко зберігається і поширюється в породі недолік, який визначається рецесивним геном, особливо якщо носієм його є популярний племінний пес, який за все життя може дати кілька сотень нащадків. Навіть якщо він несе лише один рецесивний ген, що визначає серйозний недолік, зрозуміло, не проявляючи цей недолік сам, він може швидко поширити цей недолік в породі. На той час, коли це виявиться, вже неможливо буде що-небудь виправити, особливо якщо нащадки цього пса використовувалися широко.
Коли при розведенні собак прагнуть до екстремальних характеристикам, майже обов'язково відбувається погіршення породи. Собаки тих порід, які, незважаючи на втручання людини, залишаються близькими за конституцією до їх диким предкам, які не дуже страждають від різних дефектів, закріплених в інших породах як відмітна ознака. Якби собаківники усвідомлювали, які нещастя і страждання вони заподіюють самим собакам, не кажучи вже про оплату ветеринарних послуг по виправленню характеристик екзотичних порід, то вони вже вирішили б, що розумніше змінити стандарти. Собаківники легко перестають міркувати, коли мова йде про їх собаках, хоча вони щедрі на увагу до них, але швидко стають сліпими і чесно не помічають трагедій, які він розводять. Можна навести лише деякі приклади, повний список був би занадто довгий.
Завжди бувають важкі пологи, коли черепа і газові отвори за розмірами не відповідають один одному (наприклад, у бостон-тер'єрів і французьких бульдогів); укорочені кінцівки змінюють положення клубових кісток таза, при цьому тазовий отвір виявляється занадто низько, і, якщо у собак такої породи відвислий живіт, пологи будуть важкими (наприклад, у шотландських тер'єрів), занадто довга спина і поперек дають додаткове навантаження на хребет, що виражається в захворюванні міжхребцевих хрящів (такси, бассети); занадто короткі морди ускладнюють дихання, у цих порід дуже часто, у всякому разі частіше, ніж у інших, народжуються цуценята з "вовчою пащею" (пекінеси); перебільшено вільна шкіра утворює глибокі складки, де часто виникають попрілості, відтягнуті шкірою повіки перестають захищати очі, де вникає стійкий хронічний кон'юнктивіт (бладхаунд); занадто вузький вушний прохід і шерсть, що росте тут, створюють умови для стійких захворюванні вух (фокстер'єри, пуделі); і багато багато інших ...
На жаль, в собаківництві природі не дозволяють грати властиву їй роль, всіх слабких цуценят рятують, вигодовують, а потім ще отримують від них потомство, тоді як в природі вони повинні були б померти. Закон природи: "Виживає найбільш пристосований до життя" не можна порушувати безкарно. Добре ще, що багато собак зі спадковими дефектами безплідні або менш плодовиті, так що найстрашніші недоліки не зберігаються в породі, хоча могли б за допомогою "добрих" собаківників.
У собак, так же, як і у інших тварин, завжди правильно з'єднувати краще з кращим, тільки тоді собаківник може сподіватися на кращий результат. Є багато характеристик і ознак, які визначаються не єдиною парою генів, а великим числом генів. У хортів, наприклад, неможливо передбачити швидкість бігу, так як вона визначається комбінацією великого числа спадкових факторів, тобто немає домінантних або рецесивних ознак швидкості бігу. Однак відомо, що у деяких сімей або ліній швидкість вище, таким чином, знову спаровування кращого з кращих протягом тривалого часу, безумовно, дасть кращий.
Шляхом спостереження собаківник незабаром встановить, які гени є домінантними. Але, на мою думку, значно важливіше для собаківника знати, які гени є рецесивними, так як саме вони потребують найбільшої уваги при розведенні. На жаль, не всі ознаки визначаються однаково для всіх порід домінантними або рецесивними генами. Це особливо відноситься до забарвленню шерсті.
Може бути, варто ще раз повторити, що якщо у цуценяти є ознаки, непритаманні жодному з його батьків, то ці ознаки контролюються рецесивними генами.
Тут корисно привести перелік найбільш часто зустрічаються ознак, які визначаються рецесивними генами для більшості порід:
перекус
недокус
Маленькі вуха
коротка морда
Стоячі вуха (для більшості порід)
Світла мочка носа
Світлі очі
Довжина кінцівок (для більшості порід)
Довга шерсть (рецесивна для гладкошерстних)
Гладка (коротка) шерсть (рецесивна для довгошерстих)
М'яка шерсть (рецесивна для жесткошерстних)
Пряма шерсть (рецесивна для кучерявого)
Багато з важких спадкових вад також контролюються генами, які є рецесивними для більшості порід, наприклад "заяча губа", "вовча паща", перекручений хвіст, глухота, вроджена грижа, прибулі пальці, боязнь різких звуків, сечовипускання при порушенні, заворот століття, виворіт століття, альбінізм, схильність до утворення каменів в сечовому міхурі, гемофілія, потовщення і загортання губ, катаракта та ін.
Забарвлення шерсті і пігментація для багатьох порід жорстко закріплені вимогами стандарту. Ці ознаки одні з найбільш складних з точки зору успадкування, так як контролюючі гени можуть бути домінантним для однієї породи і рецесивні для іншої, контролюючи при цьому один і той же забарвлення. Що стосується пігментації, то у всіх порід коричнева або світла мочка носа, світлі очі, загальне ослаблення пігментації визначаються рецесивними генами.
Якщо розведення потрібно вести по забарвлень, потрібно зробити перелік всіх рецесивних забарвлень, відомих в даній породі. Для тих, хто дійсно цікавиться розведенням по забарвлень, я наполегливо рекомендую книгу Кларенса Літла "Наследуемость забарвлень вовни у собак"
© Х.Хармар "Собаки і їх розведення"